I’ve been using BWA MEM a lot recently for mapping short reads from whole genome sequencing or HiC experiments. Mapping high coverage sequencing data is a time-consuming task. The easiest way to accelerate, especially on servers, is to use multiple threads which is done by adding the -t flag. I’ve seen my labmates use 20 threads, 32 threads, or 40 threads. There seem to be no offical suggestions on the number of threads that would make the mapping fastest. One natural idea is that more threads should always be faster, but is that really true? To find out the answer, I did this little experiment.
I subsampled 20 million reads from a HiC datasets of human ESC with a total reads of 320 million. The read length is 36bp. It might be better to use BWA ALN than MEM for reads of this size. But It’s Okay since what I really care about here is speed rather than accuracy. I mapped the 20 million reads to human genome reference version GRCh38 with threads of 1, 2, 4, 6, …, 100 (basically one and all even numbers within 100).
Below is the result I got. I plotted number of threads versus log2 transformed run time in minutes.
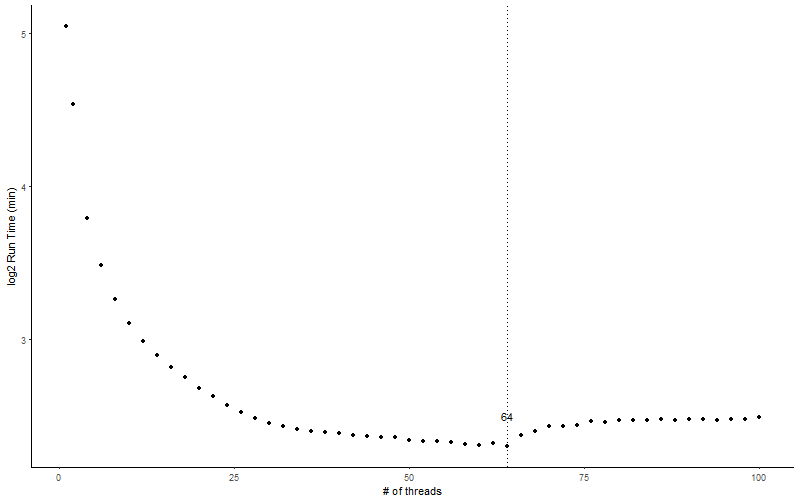
Surprisingly, it’s not the more the better. The total run time decreases rapidly from 1 thread to 2 thread, and 2 thread to 3 thread, but begins to stabilize after 10 threads until it reaches its minimal at 64 threads and then starts to increase. Even though the run time from 25 to 100 threads are roughly the same, we can still see the miminal run time is reached at 64 threads. It is imaginable that with more reads to be mapped, the speed to stablize will slow down.
The reason for minimal run time at 64 threads may be that the total threads on the server/node that BWA was run on is just 64. When adding more threads, because the CPU has sort of saturated, the computation speed will not increase.